
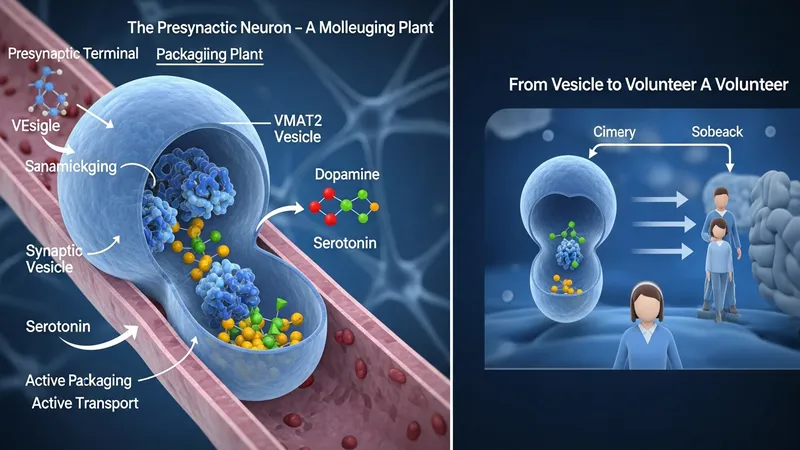
Clinical investigation of compounds that modulate vesicular monoamine transporter 2 (VMAT2) focuses on how altering presynaptic monoamine packaging may affect clinical symptoms and biological measures. Trials in this area examine investigational or approved molecules to characterize pharmacokinetics, pharmacodynamics, safety, and symptom-related endpoints. Study protocols commonly define specific populations, dosing regimens, comparator groups, and standardized outcome measures to support scientific inference rather than to promote products.
Such studies commonly progress through early-phase safety and dose-finding work into larger randomized, controlled efficacy trials and longer-term safety or post‑marketing studies. Designs may include single-ascending-dose and multiple-ascending-dose cohorts, randomized placebo-controlled arms, crossover comparisons, or adaptive features that refine doses based on interim data. Protocols also typically specify laboratory monitoring, adverse-event reporting, and criteria for stopping or modifying treatment.
Study design frameworks for VMAT2 trials may vary according to the development objective. Early-phase studies often emphasize pharmacokinetics and dose-response relationships; these may enroll small cohorts and use intensive sampling. Larger phase II or III trials may use parallel-group randomized designs with placebo or active comparators and prespecified primary endpoints. Adaptive elements such as interim sample-size re-estimation or dose-selection may be included, though these require predefined rules and regulatory consultation in the United States.
Endpoint selection typically aligns with the targeted clinical condition and regulatory expectations. For movement-related outcomes, investigator-rated scales such as the Abnormal Involuntary Movement Scale (AIMS) and clinician-rated severity measures are commonly used in US trials. Secondary endpoints can include patient-reported outcome measures, functional assessments, and objective laboratory or biomarker measures. Safety endpoints usually cover adverse event frequency, laboratory abnormalities, and measures relevant to monoamine modulation such as mood assessments.
Participant selection and eligibility criteria often balance internal validity and generalizability. Inclusion criteria may set diagnosis confirmation, severity thresholds, and stabilized concomitant medications, while exclusion criteria may screen for conditions that increase safety risk or confound outcomes. US protocols commonly require informed consent reviewed by an institutional review board (IRB) and specify safety monitoring plans including periodic laboratory testing, ECGs when indicated, and clear reporting pathways for serious adverse events.
Regulatory and transparency components are integral to trial conduct in the United States. Sponsors typically submit an investigational new drug (IND) application to the US Food and Drug Administration (FDA) before initiating many clinical studies, and trials conducted in the US are commonly registered on ClinicalTrials.gov. Institutional review and data monitoring committees may be used to oversee safety, and sponsors often plan for data submission that aligns with FDA guidance on efficacy and safety evidence.
In summary, clinical research on VMAT2-modulating compounds involves staged study designs, clearly defined endpoints, and regulatory and ethical oversight tailored to safety and scientific objectives. Protocols may vary according to phase and indication, and US-based studies commonly use established rating scales, IRB review, and trial registration. The next sections examine practical components and considerations in more detail.
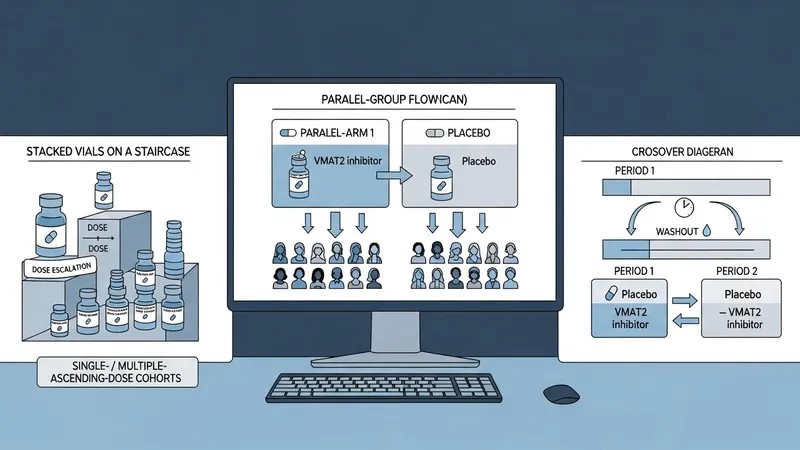
Randomized, placebo-controlled parallel-group trials are commonly used to test efficacy hypotheses for VMAT2‑active compounds in the United States. These designs allocate participants to investigational drug or placebo arms and use prespecified primary endpoints assessed at fixed time points. Crossover designs may be chosen when within-subject comparison reduces variability and when the condition and drug kinetics allow washout periods. Early-phase designs in the US often emphasize single- or multiple-ascending-dose cohorts to define tolerability and pharmacokinetics before larger controlled trials proceed.
Adaptive designs may be considered to improve efficiency; examples include dose-selection adaptations and interim futility monitoring. In the US regulatory context, adaptive elements usually require detailed pre-specification and statistical control of type I error; sponsors often discuss adaptive plans with FDA during development. Open-label extension studies are another common design to collect longer-term safety and tolerability data after controlled phases conclude.
Platform or master-protocol approaches are less common for VMAT2 programs but may be used in broader neuropsychiatric research networks in the United States to evaluate multiple agents under a shared infrastructure. These approaches can standardize outcome measures and reduce redundancy in site activation. Operational considerations for any of these designs include site experience with neuropsychiatric scales, training for raters, and clear procedures for randomization and blinding to maintain data integrity.
When determining a design, sponsors and investigators in the United States typically weigh statistical power, enrollment feasibility, and endpoint reliability. Sample-size assumptions often rely on prior US-based studies or pooled estimates from the literature. Considerations such as expected dropout rates, concomitant medication prevalence, and variability of key outcome measures in US populations are commonly incorporated into protocol planning to ensure interpretable results.

Primary endpoints in US trials are selected to reflect clinically meaningful change within the target population and to align with regulatory expectations. For movement-disorder-oriented indications, instruments such as the Abnormal Involuntary Movement Scale (AIMS) or clinician-rated severity scales are frequently used. These scales have established scoring methods and inter-rater training programs that US sites may implement to improve measurement consistency across investigators and centers.
Secondary endpoints often include patient-reported outcomes that capture perceived functional impact, quality of life, or specific symptom dimensions. Objective measures such as timed tasks, digital phenotyping, or laboratory biomarkers may supplement clinical scales to provide convergent evidence. In US studies, sponsors typically describe how each endpoint will be analyzed, how missing data will be handled, and which endpoints are considered key to benefit-risk assessment.
Safety monitoring endpoints cover adverse event rates, laboratory tests, cardiac monitoring when relevant, and psychiatric evaluations for mood symptoms or suicidality if indicated by the pharmacology. Data collection plans usually define the frequency of assessments and thresholds for action. In the United States, serious adverse events are reported through established sponsor-IRB-FDA pathways, and protocols often reference specific safety assessments required by agency guidance.
Exploratory endpoints may include neuroimaging, pharmacogenomic profiling, or other biomarkers to explore mechanism of action or predictors of response. While exploratory outcomes are not typically used for regulatory claims, US investigators often collect them to inform future development. Protocols usually specify whether such data will be analyzed centrally and how results may inform subsequent study designs.

Eligibility criteria in US protocols commonly require confirmed diagnosis using standardized diagnostic criteria and may set thresholds for symptom severity or duration. Trials often limit enrollment of participants with unstable medical comorbidities, recent use of interacting medications, or histories that could confound outcome interpretation. Inclusion and exclusion rules are designed to protect participant safety while maintaining sufficient generalizability to target clinical populations seen in US practice.
Recruitment strategies in the United States often involve site networks familiar with relevant populations, referrals from specialty clinics, and registry outreach where permitted. Recruitment plans typically include realistic timelines reflecting experience in US trials for similar indications, and sponsors usually monitor enrollment metrics to identify and address site-specific barriers. Demographic diversity considerations are increasingly described in US protocols to improve representativeness.
Safety monitoring plans detail the schedule of clinical and laboratory assessments, procedures for documenting adverse events, and rapid reporting mechanisms for serious events. US studies commonly use centralized safety reporting and may convene data and safety monitoring boards (DSMBs) for larger or higher-risk trials. Protocols also describe investigator responsibilities for local safety oversight and communication with IRBs and regulatory authorities.
Informed consent procedures in US trials include disclosure of known risks and potential benefits in neutral language, explanation of voluntary participation, and documentation processes. Many US sites use consent forms reviewed by IRBs and provide time for participants to ask questions. Ongoing safety follow-up and provisions for withdrawal or discontinuation are typically outlined to protect participant welfare and data integrity.

Sponsors conducting VMAT2-related studies in the United States commonly engage with the FDA through pre-IND or end-of-phase meetings to align on development plans, endpoints, and safety assessments. Many US trials operate under an IND when testing investigational compounds. Regulatory interactions often cover design choices, inclusion of adaptive features, and expectations for pivotal evidence. Sponsors typically document these discussions and reflect them in protocol rationales to support later regulatory submissions.
Trial registration on ClinicalTrials.gov and adherence to reporting requirements are standard for US-based studies. Operational planning also addresses site selection, investigator training for specific rating scales used in the study, and central monitoring to detect data quality or protocol deviations. Contracts, budgeting, and institutional approvals commonly reflect US institutional review processes and local site policies.
Data safety oversight in US trials may involve DSMBs, independent safety monitors, and predefined stopping rules. Sponsors often establish statistical analysis plans that prespecify primary analyses, multiplicity controls, and sensitivity analyses. Quality assurance activities such as monitoring visits, source-data verification, and audits are typically part of US operational plans to meet regulatory expectations and ensure reliable data capture.
Post-study obligations in the United States can include timely adverse-event reporting, data submission consistent with FDA requirements, and registration updates on ClinicalTrials.gov. Sponsors and investigators may also plan for dissemination of results in scientific forums and for data-sharing discussions, always ensuring protection of participant confidentiality and adherence to applicable US regulations governing human subjects research.