
Tirzepatide injections are an injectable pharmaceutical intervention that acts on multiple incretin receptors to influence metabolic pathways. In clinical and regulatory descriptions, this compound is classified as a dual glucose‑dependent insulinotropic polypeptide (GIP) and glucagon‑like peptide‑1 (GLP‑1) receptor agonist. In practice, discussions about such injections typically cover the biological mechanism, how dosing is scheduled and adjusted, and safety considerations that clinicians and patients commonly review together. Descriptions emphasize possible effects on appetite, glucose regulation, and gastrointestinal function while noting that individual responses may vary.
When examining these injections from a dosing and safety perspective, key components include typical administration frequency, sender device formats, dose‑titration strategies, and monitoring for adverse effects. Health professionals often consider existing clinical trial data, product labeling, and patient comorbidities when discussing options. The following list gives concise examples of items or comparators that are relevant in clinical and educational contexts about this class of injectable therapies.
Mechanistic descriptions commonly note that a dual GIP/GLP‑1 receptor agonist may interact with receptors involved in insulin secretion and appetite regulation, and that these interactions can influence several physiological processes. Pharmacokinetic properties such as absorption after subcutaneous injection and a multi‑day elimination profile are typically presented in product documentation. Clinical study reports commonly provide averaged outcomes for groups rather than promises about single patients; therefore, descriptions in educational materials remain framed in terms such as may, can, or typically.
Typical dosing approaches described in labels and clinical summaries often include an initiation dose followed by stepwise titration to reach a maintenance dose, with weekly dosing schedules in many formulations. Titration is commonly intended to balance potential benefits with tolerability, especially for gastrointestinal symptoms that are frequently reported in trials. Documentation also outlines contraindications, precautions, and scenarios warranting specialist consultation or dose adjustments.
Conversations about safety considerations usually prioritize known patterns of adverse effects, monitoring needs, and the importance of reviewing concomitant medications. Product labels and clinical reviews characterize common reactions and rare but serious events, recommending targeted surveillance for symptoms that may require clinical assessment. Educational materials stress shared decision‑making and individualized assessment rather than universal recommendations.
Contextual comparisons often place tirzepatide within a broader group of incretin‑based injectables, highlighting differences in receptor profiles, dosing frequency, and trial endpoints used in regulatory filings. Comparative data in regulatory summaries and peer‑reviewed literature typically use cautious language about relative effects, reporting averaged group results instead of definitive statements about superiority. Readers are encouraged to interpret comparisons as part of a broader clinical picture where multiple variables may influence outcomes.
Regulatory documents and clinical trial reports provide the main evidence base for dosing schedules and labeled safety information; these sources often include population characteristics, common adverse reactions, and recommended monitoring. When interpreting these sources, it is typical to consider trial inclusion criteria, background therapies, and the follow‑up duration, as these factors can affect how applicable trial findings are to individual clinical settings. Educational summaries generally reflect these contextual limits and encourage further discussion with qualified clinicians.
In summary, an educational overview of tirzepatide injections centers on mechanism, administration patterns, dose‑titration strategies, and safety and monitoring considerations as described in product documentation and clinical literature. Information is typically presented with cautious language and reference to authoritative sources. The next sections examine practical components and considerations in more detail.
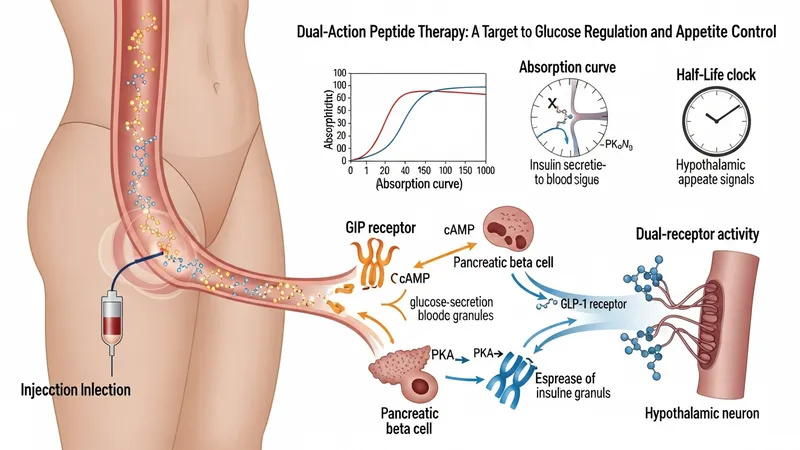
Descriptions of the pharmacological actions emphasize that the agent engages both GIP and GLP‑1 receptor pathways, which may influence insulin secretion and appetite signals in a glucose‑dependent manner. Product documentation explains receptor affinities, downstream signaling effects, and how these properties relate to observed clinical outcomes. Pharmacology sections also cover absorption characteristics after subcutaneous injection and typical half‑life estimates used to inform dosing frequency. These mechanistic summaries are used to explain rationale rather than to promise specific individual outcomes.
Clinical literature often details how dual‑receptor activity differs from single‑receptor GLP‑1 agonists in study endpoints and biomarker changes; however, such comparisons are typically framed as observational or hypothesis‑generating rather than conclusive. Pharmacodynamic data presented in trials may show averaged changes in metabolic markers across study populations. Educational material will note that individual variability—due to genetics, concurrent medications, or comorbid conditions—can influence how a person responds pharmacologically.
Safety and pharmacology sections also discuss potential interactions with other medications affecting glucose metabolism, gastric emptying, or renal function. Product labeling commonly provides lists of drugs where monitoring or dose adjustments could be considered. When covering these interactions, neutral phrasing is used to indicate that clinicians may need to review the full medication profile and laboratory data to assess relevance for a specific patient.
Summaries of mechanism and pharmacology in patient‑oriented materials generally avoid technical overload while highlighting the practical implications for dosing intervals and anticipated tolerability patterns. These summaries often recommend consulting prescribing information or professional guidance for detailed pharmacokinetic parameters and caution that trial conditions may differ from routine clinical practice. Continued reading will address dosing basics and administration considerations.
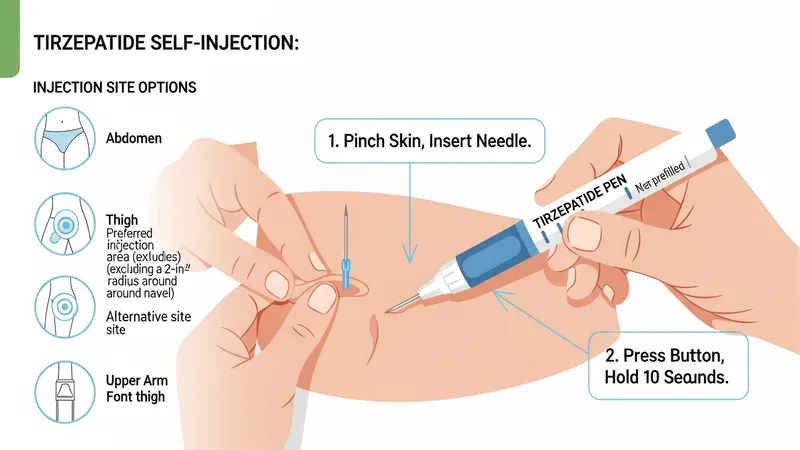
Product labeling commonly outlines an initiation dose, stepwise titration increments, and a maintenance dose range for approved indications. Typical schedules often use weekly subcutaneous injections delivered via prefilled pen devices. Educational descriptions emphasize that titration is intended to improve tolerability and that dose adjustments may be advised for adverse reactions or concomitant medical conditions. All dosing examples in this content are descriptive summaries; prescribing information is the definitive source for specific regimens.
Administration guidance in official materials covers injection technique, storage of prefilled devices, and handling precautions. Many formulations are designed for subcutaneous administration in areas such as the abdomen, thigh, or upper arm, and labeling commonly addresses needle use, disposal, and what to do if a dose is missed. These operational details are presented to inform discussions between patients and clinicians about practical aspects of treatment.
Clinical summaries often note that dosing decisions consider patient factors such as renal function, gastrointestinal history, or prior exposure to incretin‑based therapies. In some studies, prior use of GLP‑1 receptor agonists was accounted for in subgroup analyses. Educational materials advise that clinicians may tailor titration speed and maintenance targets based on individual tolerability and clinical context, using cautious language rather than prescriptive statements.
When discussing dosing in broader contexts, materials frequently highlight the importance of reviewing official labeling for contraindications, warnings, and dose modification guidance. Health professionals may also reference contemporary clinical guidance documents or systematic reviews for context on real‑world adherence patterns and device usability. The following section will examine safety considerations and common adverse events.
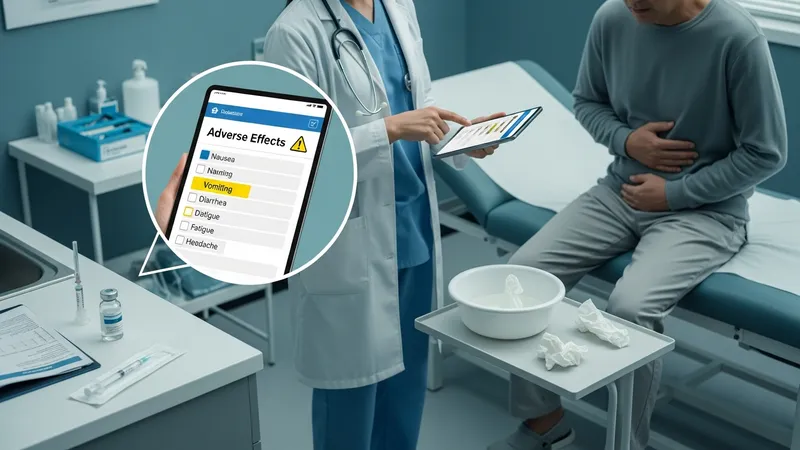
Safety summaries for these injections generally report a pattern of gastrointestinal adverse effects as commonly observed in clinical trials, with nausea, vomiting, and diarrhea among the more frequently cited events. Product documentation also lists less common but potentially serious events that warrant clinical attention. Educational content uses measured language to indicate that frequency and severity of adverse effects may vary across individuals and trials, and that monitoring plans are often recommended in labeling and clinical guidance.
Labels and clinical reviews typically describe rare events or conditions included as warnings, and they often recommend specific actions if certain symptoms occur. For example, guidance may suggest evaluation for severe persistent gastrointestinal symptoms or signs that could indicate a more serious complication. Educational materials emphasize that such information is intended to inform monitoring strategies rather than to imply inevitability of these outcomes.
Considerations often extend to comorbid conditions and concurrent medications that may alter risk profiles. For instance, patients with a history of pancreatitis or specific endocrine neoplasms may need individualized assessment; product labels commonly direct clinicians to review these histories before initiating therapy. Safety discussions present these scenarios as considerations for clinical judgment rather than as absolute contraindications applicable to all patients.
Post‑marketing surveillance and longer‑term observational studies are often referenced to describe how safety profiles evolve beyond clinical trials. These sources may report incidence patterns over larger and more varied populations, but they are typically characterized by cautious phrasing about association versus causation. The next section will address monitoring and communication elements relevant to clinical follow‑up.

Monitoring recommendations found in product information and clinical guidance commonly include baseline assessments and periodic follow‑up for metabolic parameters, renal function, and symptom review. Lab tests and clinical evaluations are described as tools to assess therapy tolerability and to detect potential adverse events that may necessitate dose modification or discontinuation. Educational materials typically frame monitoring as a routine component of shared decision‑making between clinician and patient.
Clinical follow‑up strategies often emphasize reviewing response, side effects, and adherence to the prescribed dosing schedule. Shared decision‑making frameworks presented in professional guidance suggest discussing expectations, possible adverse effects, and how to manage common symptoms. Informational resources encourage documenting changes and reassessing goals over time as part of an ongoing clinical plan rather than as a one‑time directive.
Healthcare providers may use existing clinical checklists or electronic health record reminders to track relevant parameters and ensure appropriate follow‑up intervals. Observational evidence and registry data sometimes inform recommended monitoring frequency, but such recommendations are usually presented as flexible and adaptable to patient needs. Educational content will note that individualized follow‑up plans are typical in routine practice.
Concluding discussions in monitoring sections advise that patients and clinicians consult authoritative product labeling and specialty guidance for specific monitoring recommendations, and that any decision about initiation or continuation of therapy should be based on individualized clinical assessment. These points are framed as informational considerations rather than prescriptive actions.